Urgent Message: Progressive diaphyseal dysplasia, also known as Camurati-Engelmann disease, is a rare genetic disease that predominantly affects the bones. Clinicians who understand the manifestations of this disorder are better equipped to ensure appropriate management of disease flares and coordinate specialist follow-up.
Swetha Gogu, DO, MPH; Sudhir Gogu, DO, PhD, MBA
Citation: Gogu S, Gogu S. Progressive Diaphyseal Dysplasia: A Case Report. J Urgent Care Med. 2024; 18 (11): 34-37
Abstract
Introduction: Camurati-Engelmann disease (CED), also known as progressive diaphyseal dysplasia, is a rare autosomal dominant genetic disease that predominantly affects the bones.
Presentation: A 30-year-old male presented to the urgent care (UC) in a wheelchair with acute on chronic weakness in his upper and lower extremities. He also endorsed pain in his extremities for several days.
Diagnosis: The diagnosis of CED is often made from clinical and radiological findings. However, given that it is an autosomal dominant disease, molecular genetic testing for mutations in transforming growth factor beta-1 (TGFB1) can confirm the diagnosis.
Resolution: The patient was prescribed a course of corticosteroids for the acute pain. Referrals to orthopedics, genetics, endocrinology, and physical therapy were made for further evaluation and management.
Conclusion: CED is a rare form of skeletal dysplasia. It is important for UC providers to understand the manifestations of patients with this disorder and ensure appropriate specialist follow-up for this chronic, debilitating disorder.
Introduction
Camurati-Engelmann disease (CED), also known as progressive diaphyseal dysplasia, is an ultrarare autosomal dominant disease. There have been only about 300 total cases identified. The disease typically presents in childhood but may not present until late into adulthood for some individuals. Patients most often initially present with complaints of limb pain and fatigue. Patients often have a characteristic waddling gait. CED occurs when a dysfunctional copy of the gene encoding TGF-β is present. It is characterized by hyperostosis of the long bones and skull as well as severe bone pain with consequent weakness and gait alterations.[1],[2]
Clinical Presentation
A 30-year-old male presented to an UC center in a wheelchair with complaints of 3 days of weakness in his upper and lower extremities. He also endorsed pain in his lower extremities and difficulty walking for several days. He denied paresthesias, headaches, hearing loss, or vision changes. His first medical evaluation for lower extremity weakness and pain was in his mid-teens, at which time he was diagnosed with CED. He had previously been managed with occasional courses of corticosteroids or non-steroidal anti-inflammatory drugs (NSAIDs) by his primary care physician (PCP), but because he was unable to see his PCP, he sought pain relief for his current pain flare at the UC center.
Physical Exam and Findings
The patient’s vitals at the time of his UC visit were within normal limits. On physical exam, he appeared uncomfortable due to pain. His cardiopulmonary and abdominal exam were within normal limits. His musculoskeletal exam revealed atypically elongated extremities with lumbar lordosis and mild swelling of bilateral ankles. Due to pain, his active and passive range of motion was limited in upper and lower extremities. His neurologic exam revealed symmetrically diminished strength in all muscle groups of all 4 extremities. His sensation was intact to light touch throughout, and his deep tendon reflexes were normal.
Differential Diagnosis
CED has characteristic clinical and radiological findings, however, it is important to avoid anchoring on this diagnosis prematurely in patients with CED presenting with acute limb pain. Many clinical entities were considered as possible explanations for the patient’s acute pain and weakness including infection (eg, osteomyelitis) and oncologic processes involving bony metastases. Co-existent chronic bone disease such as Ribbing’s disease (multiple diaphyseal sclerosis), Paget’s disease, Kenny-Caffey syndrome, osteopetrosis, avascular necrosis, and osteosclerosis were also considered. Neuropathies and myopathies can cause pain and weakness, but given the patient’s description of the symptoms resembling prior episodes, it was felt that this case was most likely related to exacerbation of CED.
Urgent Care Management and Diagnostic Assessment
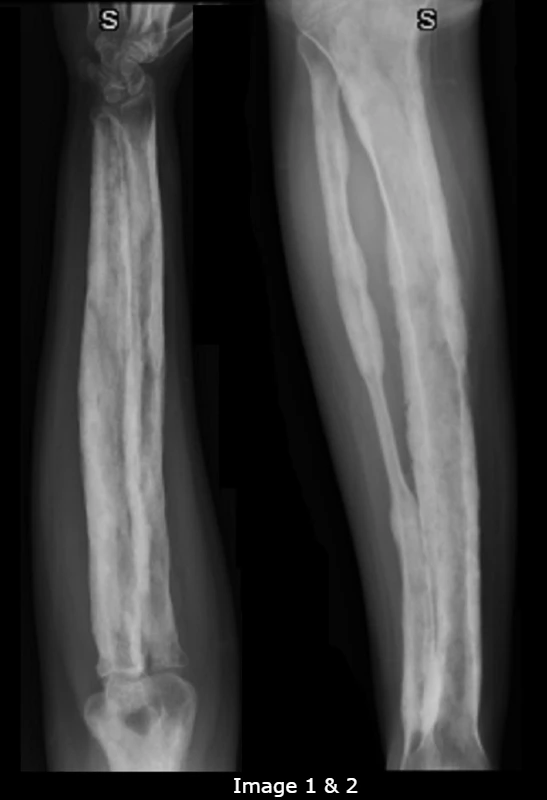
X-ray (XR) images of the bilateral lower legs and forearms were obtained to assess for progression of disease and alternate pathologies (Images 1-2). The radiologist’s interpretation of the XR suggested that there was symmetric, irregular cortical thickening and periostitis of the long bones. There were no fractures or osseous lesions. As there are no diagnostic criteria for CED, the patient’s diagnosis of CED in his mid-teens was presumed, based on his known history and associated radiographic findings.
Case Conclusion
The patient was prescribed a course of oral corticosteroids and advised to avoid NSAIDs while taking the steroids. Referrals to orthopedics, genetics, endocrinology, and physical therapy were made for further treatment and management. Additional recommendations included vision evaluation, and the patient was provided anticipatory guidance on expected course of the disease.
Epidemiology
CED was first described in 1920.[3] Rarely, the disease can come from a spontaneous genetic mutation in the egg or sperm cell. The prevalence of CED is unclear, but there have been over 300 cases reported to date worldwide.[4]
Pathophysiology
CED is a rare skeletal disorder that belongs to the group of sclerosing bone dysplasias caused by mutations in the Transforming Growth Factor Beta 1 (TGFB1) gene. The gene is located on chromosome 19q13. It encodes the TGFβ-1 protein, which is found throughout the body but is particularly prevalent in the skeletal system where it helps regulate the formation and growth of bone and cartilage. The TGFB1 gene mutations that cause CED result in the production of an overly active TGFβ-1 protein. The abnormal activity in this protein causes an increase in signaling, which leads to more bone formation. As a result, the bones in the arms, legs, and skull are thicker than normal, contributing to the movement and neurological problems often experienced by individuals with CED.[5]

Presentation
Patients generally present with pain in the extremities, decreased muscle mass and symmetric proximal muscle weakness, contractures, wide-based waddling gait, bone pain, frontal bossing, and easy fatigability. If bones of the skull are affected, then individuals may experience headaches, hearing loss, tinnitus, vertigo, vision problems, and even facial paralysis if the nerves become compressed. Some individuals may also present with abnormally long limbs in proportion to the height of their body, a decrease in muscle mass and body fat, visible prominence of the long bones in the legs, and rarely delayed puberty. Pain symptoms tend to be exacerbated with cold, stress, and increases in activity.1,4,[6]
Treatment
There are currently no disease modifying treatments available for CED. Corticosteroids are reported to help relieve the symptoms during acute flares but do not slow progression of the disease.1 The use of steroids must be weighed against the long-term risks they may pose. Additionally, there are limited case reports of losartan reducing bone pain and increasing physical activity as it has been shown to downregulate TGFB1 signaling.[7] There is limited evidence with use of bisphosphonates.2 NSAIDs and other analgesics and non-pharmacological (eg, heat or ice) methods can be used to treat the pain. Selective cases may benefit from surgical intervention. Often a multidisciplinary team of physical therapists, occupational therapists, and physiatrists is important to help manage long term quality of life.7
Patients with CED are most likely to present to UC during acute pain exacerbations. In these instances, it is appropriate to treat pain symptomatically. However, it is important to ensure such patients have appropriate specialist care for follow-up. Most patients with CED will have 1 or more specialists involved in their care already, and it is prudent to consult with their primary specialist when feasible. Referral to a geneticist early in the course is prudent, although many patients with CED will have already undergone genetic evaluation. An orthopedist may be consulted due to concerns for bone dysplasia, and an endocrinologist to ensure proper growth and development of the bones. Additionally, patients with skull bases involvement should have routine ophthalmological, neurological, and otolaryngologic follow up.⁶
Discussion
While CED is a rare cause of skeletal dysplasia, patients with this condition may present to UC with exacerbations in pain and/or weakness. As the timing of diagnosis is variable, it is important to consider CED in the differential diagnosis for patients presenting with non-specific limb pains and radiological features of skeletal dysplasia. The prognosis for individuals with CED is highly variable and depends on the severity of the disease and the presence of complications. The condition is typically progressive, with symptoms worsening over time.4,6
Ethics Statement
An attempt was made to contact the patient to obtain informed consent to publish this case, but he was lost to follow-up and could not be reached. The patient’s identifying details were changed or omitted to protect patient privacy.
Takeaways for Urgent Care Providers

- CED is diagnosed primarily on the basis of history and radiologic examination, however confirmation can be made with genetic testing.
- The most common manifestations include bony pain of the extremities and proximal muscle weakness. The disease usually becomes clinically apparent early in life, in which most patients are diagnosed.
- There are currently no disease modifying treatments available for CED, however, short courses of oral corticosteroids have shown some efficacy in reducing exacerbations of pain.
- UC clinicians should make efforts to consult with specialists who are familiar with the CED diagnosis when making UC treatment decisions in order to coordinate appropriate follow-up.
Manuscript submitted April 29, 2024; accepted August 6, 2024.
References
- [1]. Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, Guanabens N, Migone N, Wientroub S, Divizia MT, Bergmann C, Bennett C, Simsek S, Melancon S, Cundy T, Van Hul W (2006) Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet 43:1–11
- [2]. Wallace SE, Lachman RS, Mekikian PB, Bui KK, Wilcox WR. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati-Engelmann disease): report of a four-generation pedigree, identification of a mutation in TGFB1, and review. Am J Med Genet A. 2004;129A(3):235 247.
- [3]. Klemm P, Aykara I, Lange U. Camurati-Engelmann Disease: A Case-Based Review About an Ultrarare Bone Dysplasia. Eur J Rheumatol. 2023;10(1):34-38. doi:10.5152/eurjrheum.2023.21115
- [4]. Sparkes RS, Graham CB. Camurati-Engelmann disease. Genetics and clinical manifestations with a review of the literature. J Med Genet. 1972;9(1):73-85. doi:10.1136/jmg.9.1.73
- [5]. Camurati-Engelmann Disease – NORD (National Organization for Rare Disorders). NORD (National Organization for Rare Disorders). Published 2019. Available from: https://rarediseases.org/rare-diseases/camurati-engelmann-disease/
- [6]. Wallace SE, Wilcox WR. Camurati-Engelmann Disease. 2004 Jun 25 [Updated 2023 Aug 31]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1156/
- [7]. Das L, Dhiman V, Dutta P, et al. Camurati-Engelmann Disease Complicated by Hypopituitarism: Management Challenges and Literature Review of Outcomes With Bisphosphonates. AACE Clin Case Rep. 2021;8(2):58-64. Published 2021 Oct 20. doi:10.1016/j.aace.2021.10.002
Download the article PFD: Progressive Diaphyseal Dysplasia: A Case Report